Research Overview:
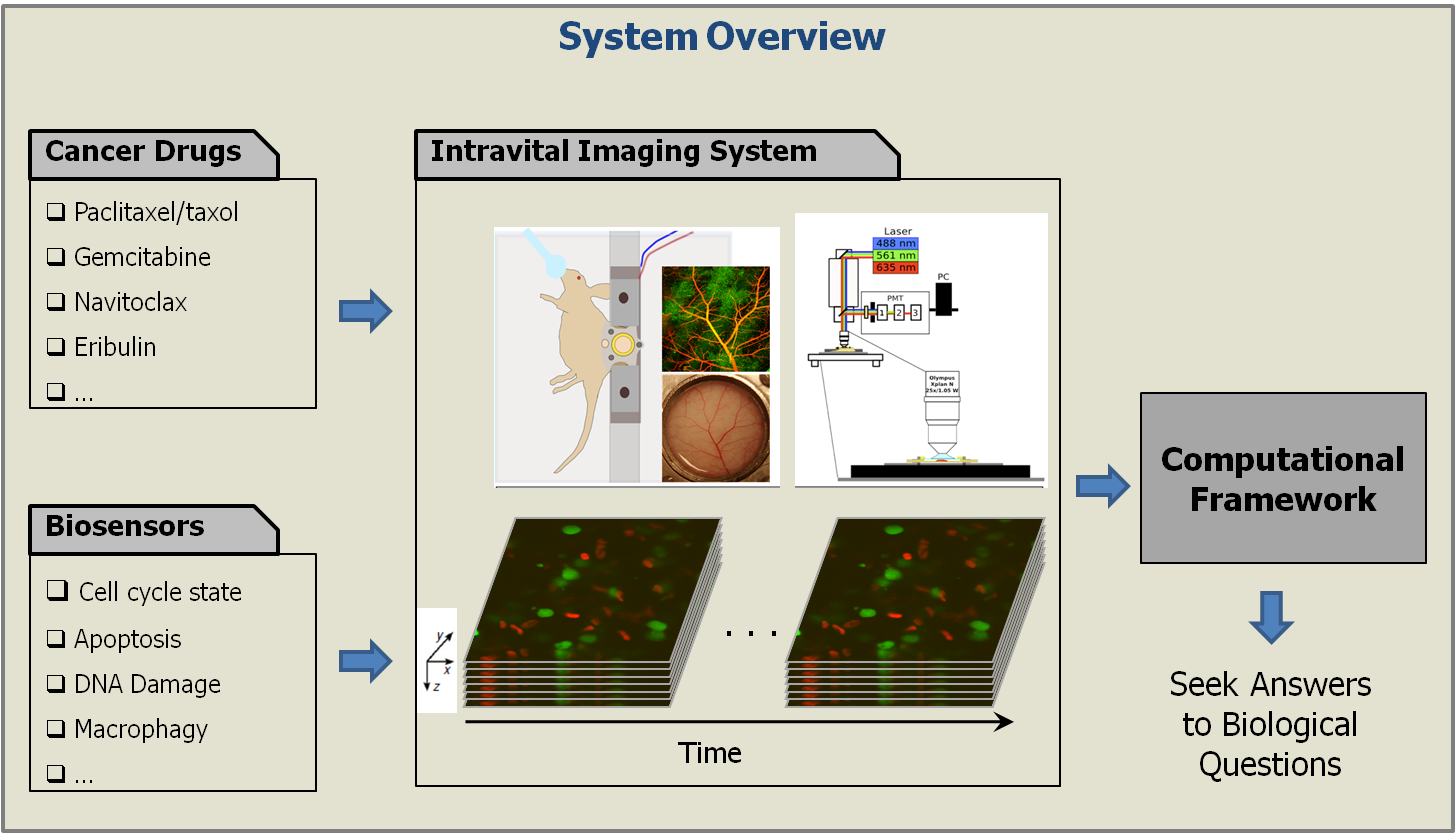
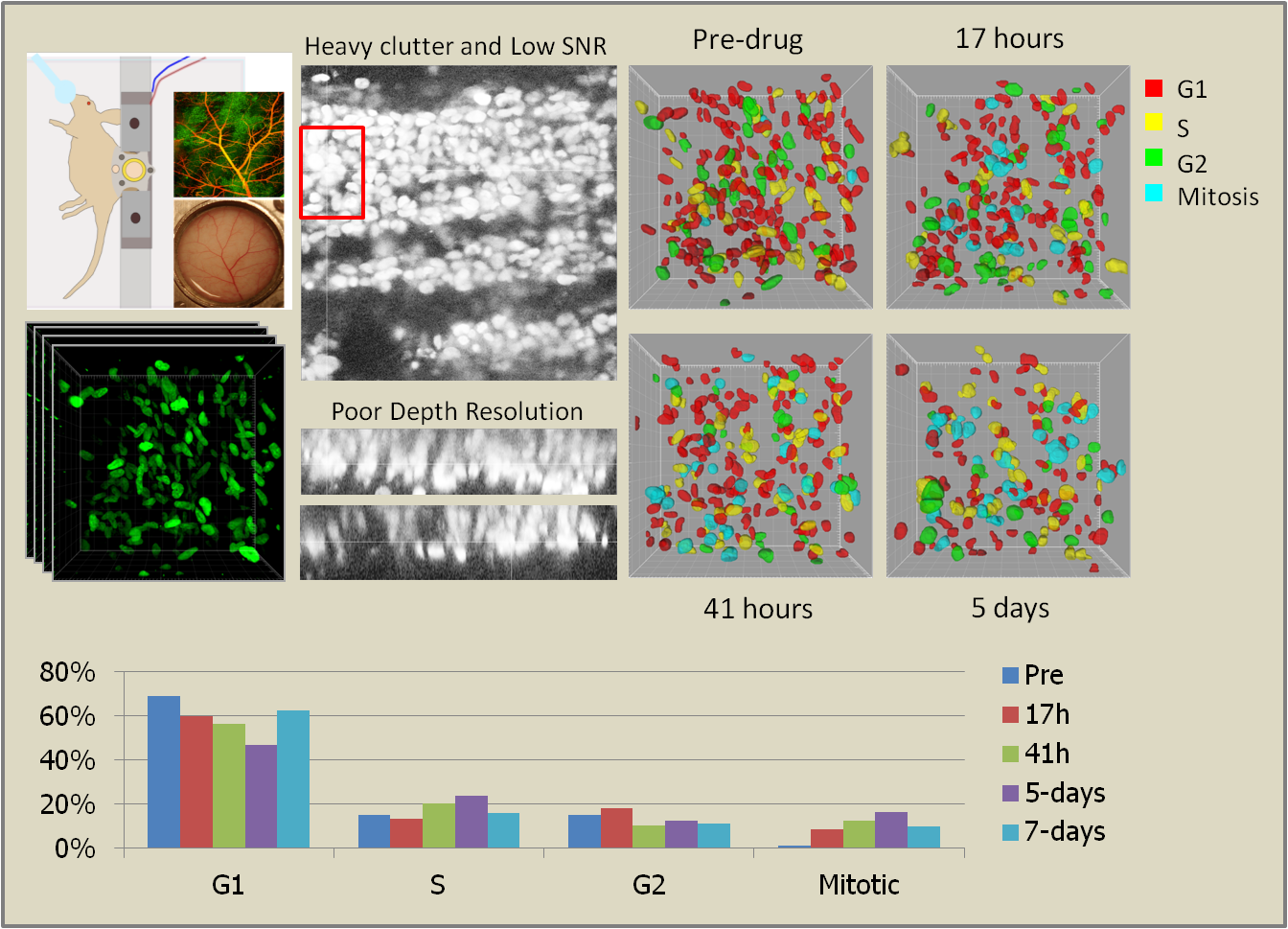
Cancer continues to be one of the leading causes of death worldwide and imaging, both at the microscopic and macroscopic scales, continues to serve as an invaluable tool in all facets of cancer research including diagnosis, clinical treatment and drug discovery. Microscopic imaging systems, in particular, have been very useful in gaining a deeper understanding of cancer cell biology thereby facilitating the design and discovery of better anti-cancer drugs. However, the bulk of cancer biology has been conducted using in vitro and ex vivo methods, neither of which provide an accurate representation of the complex in vivo processes. To better understand the origin and progression of cancer and its response to chemotherapeutic drugs, we need to image cancerous tissue in live animals at a microscopic resolution, a technique popularly known as intravital microscopy. However, intravital images present a whole new challenge for computational image analysis including low foreground-background contrast, weak edge information between adjacent objects, high morphological complexity, and poor depth resolution. Todays best practice in intravital imaging is anecdotal observations of select image events. This, however, is insufficient to draw reliable inferences about the mechanisms of chemotherapeutic drug action. We have begun to tackle these image analytical challenges in a project aiming at the quantification of drug action at the single cell level in human tumor models cultured in living mice imaged intravitally in 3D using laser scanning fluorescence microscopy.
To this end, we have developed a computational framework for the automated quantification and monitoring of cell cycle state distribution at multiple time points before and after treatment with cancer drugs. This framework is the key facilitator for data analysis in all experimental work performed in this work by (i) largely automating the analysis of the generated microscopy data thereby enabling studies at a much larger scale, and (ii) ensuring reproducibility and cross-experiment consistency of analysis unattainable by manual quantification. From a theoretical point of view, we have developed algorithms for robust segmentation of hundreds of cells embedded in the highly cluttered tumor environment followed by their association to specific states of the cell cycle. Next, we will develop statistical methods to test biological hypotheses regarding the dynamics and spatial patterning of drug responses.
Team Members:
Deepak Roy Chittajallu and Gaudenz Danuser, Laboratory of Computational Cell Biology, Harvard Medical School
Stefan Florian and Tim Mitchison, Mitchison Lab, Harvard Medical School
Rainer Kohler and Ralph Weissleder, Weissleder Lab, Massachusetts General Hospital
Lee Albacker and Peter Sorger, Sorger Lab, Harvard Medical School